Histopathology
This page describes the algorithm and the use of the program for nucleus and whole cell segmentation and nucleus/cellular scale biomarker quantification. The algorithms have been implemented in c++ and IDL and instructions to use the program are also included.
Contents |
Background and motivation
Detecting molecules of interest and quantifying them plays an increasingly important role in modern medical diagnosis and treatment planning[1]. Currently, most systems are visually based qualitative descriptors which suffer from the consequent limitations. There are other algorithms that quantify biomarkers on a pixel-by-pixel basis or a regional basis[2-5]. Segmenting the cells and nuclei allows us to classify the different cell types based on antigens that mark specific cell. This enables us to quantify the biomarker of interest in the relevant cell type. This is a more biologically relevant measure and can be more useful in medical diagnosis.
Reminder: Scale and add images to show multiplexed and unmixed histological samples
Algorithm
The algorithm involves the following steps:
- Segment(delineate) nuclei
- For this we use the 2-D nucleus segmentation algorithm described here[[1]]
- Delineate cell boundaries
- If reliable membrane or cytoplasm staining is available then:
- Binarize the image by graph cuts[6]
- Create a gradient enhanced distance map with the segmented nuclei as the initialization points ( described in manuscript which will soon be submited )
- Use a seeded watershed algorithm with the gradient weighted map as the feature input and the segmented nuclei as the markers and segment(delineate) the cells[7]
- If neither the cytoplasm nor the membrane staining is reliable then use a purely geometric approach to estimate the cytoplasmic domains:
- Draw dynthetic boundaries by redefining the dominance region described by Nath,et. al.[8] as
- Draw dynthetic boundaries by redefining the dominance region described by Nath,et. al.[8] as
- If reliable membrane or cytoplasm staining is available then:
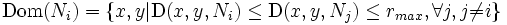
- where (x,y)is any point in the neighborhood of the nucleus Ni and rmax is the user defined maximum radius of of the synthetic boundaries. rmax is in terms of the number of pixels from nucleus Ni
- Quantify biomarker in the region of interest
- Load training set and classify cells as biomarker positive or negative using a Bayesian classifier
Histopathology Program
The algorithms have been implemented with a custom GUI built on IDL and c++. To run the program, you will need the IDL virtual machine which is available online for free[[2]]. Soon, the code will be translated into c++ and added to the Farsight repository.
Steps for analyzing 2-D multi-spectral images of Hisopathology samples
1- Read image channels one-by-one:
-Choose "start/end read image channels" from the Read menue -start reading the channels one by one (nuclei, cytoplasm and/or membrane channels)
<add a stcrean shot here>
2- Start nuclear segmentation (nuclear segmentation menue):
-Change the nuclear segmentation settings (if needed) -Run initial segmentation -Edit the nuclear segmentation results (optional). This includes merging, splitting, and deleting nuclei -Finalize segmentation. The segmentation result will be displayed and will be saved into a file as well -If a nuclear segmentation results file is already available, choose "Read Final Segmentation"
<add a screen shot here>
3- Start cytplasm (or membrane) segmentation (cytoplasm segmentation menue):
-Change the cytoplasm settings (if needed) -Run cytoplasm segmentation
<add a screen shot>
4- Quantify the biomarker (feature extraction menue):
-Choose "Quantify Biomarker"
-A new window will then allow you to brows for the biomarker channel (image)
-Set the quantification options:
-Define the region of interest
-Choose whether or not you need to do background subtraction (two- or three-level)
-Quantification results will be saved into a csv file
<add a screen shot here>
3- Classify cells (cell classification menue):
-Define classes -Select training samples manually or read them from a file saved after previous manual selection of training samples -Train the classifier -Classify the cells -classification results will be displayed (by changing the seeds colors) and will be also saved into a csv file
<add a screen shot>
References
- Hammerschmied CG, Walter B, Hartmann A. [Renal cell carcinoma 2008 : Histopathology, molecular genetics and new therapeutic options.]. Pathologe 2008;29(5):354-63.}
- Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med 2002;8(11):1323-7.
- Mulrane L, Rexhepaj E, Penney S, Callanan JJ, Gallagher WM. Automated image analysis in histopathology: a valuable tool in medical diagnostics. Expert Rev Mol Diagn 2008;8(6):707-25.
- Taylor CR, Levenson RM. Quantification of immunohistochemistry--issues concerning methods, utility and semiquantitative assessment II. Histopathology 2006;49(4):411-24.
- Tamai S. Expert systems and automatic diagnostic systems in histopathology--a review. Rinsho Byori 1999;47(2):126-31.
- Al-Kofahi Y, Lassoued W, Lee W, Roysam B. Improved Automatic Detection & Segmentation of Cell Nuclei in Histopathology Images. IEEE Trans Biomed Eng 2009 (in review)
- Beare R., Lehmann G. The watershed transform in ITK - discussion and new developments. The Insight Journal - 2006 January - June [[3]]
- Nath SK, Palaniappan K, Bunyak F. Accurate Spatial Neighborhood Relationships for Arbitrarily-shaped Objects using Hamilton-Jacobi GVD. Lect Notes Comput Sci 2007;4522 LNCS:421-431.