Histopathology
(→Histopathology Program in c++) |
(→Histopathology Program in c++) |
||
| Line 40: | Line 40: | ||
Images can be loaded as is from the hard disk. The supported file formats have the following extensions .tif, .pic, .png, .jpg and .lsm | Images can be loaded as is from the hard disk. The supported file formats have the following extensions .tif, .pic, .png, .jpg and .lsm | ||
Since there is no open source format with an accompanying tool with which a user can conveniently synthesize images composed of channels multiple channels and append meta-data, the histopathology tool includes a method to load channels and meta-data specified in a xml file(hotlink to scr cap of xml). The full path(hotlink to scr cap of xml) of each image has to be specified here(hotlink to scr cap of xml). The display color-space to which the image is rescaled is specified as <r="___" g="___" b="___">(hotlink to scr cap of xml) along with the channel name. The rage for each color is between 0 and 255. If r=g=b=255 the image will be loaded with its native color range but such images can not be used in any processing step. The conversion and rescaling of RGB images loaded as single channels is done internally, so, the format of the image is not a factor. The tag <chname="___">(hotlink to scr cap of xml) specifies the channel name which is used to identify the cahnnel in the processing step. Caution: The case of channel name strings have to match in both the image file and the project definition file. | Since there is no open source format with an accompanying tool with which a user can conveniently synthesize images composed of channels multiple channels and append meta-data, the histopathology tool includes a method to load channels and meta-data specified in a xml file(hotlink to scr cap of xml). The full path(hotlink to scr cap of xml) of each image has to be specified here(hotlink to scr cap of xml). The display color-space to which the image is rescaled is specified as <r="___" g="___" b="___">(hotlink to scr cap of xml) along with the channel name. The rage for each color is between 0 and 255. If r=g=b=255 the image will be loaded with its native color range but such images can not be used in any processing step. The conversion and rescaling of RGB images loaded as single channels is done internally, so, the format of the image is not a factor. The tag <chname="___">(hotlink to scr cap of xml) specifies the channel name which is used to identify the cahnnel in the processing step. Caution: The case of channel name strings have to match in both the image file and the project definition file. | ||
| − | + | [[Image::Load_multi_channel_image.jpg |thumb|600px|Click on the image to enlarge|center]] | |
===Nucleus Segmentation=== | ===Nucleus Segmentation=== | ||
===Whole Cell Segmentation=== | ===Whole Cell Segmentation=== | ||
Revision as of 18:11, 4 February 2010
This page describes the FARSIGHT approach applied to digital histopathology. In current clinical practice, histological specimens (from biopsy) are stained using traditional stains like hematoxylin and eosin, and inspected under a standard brightfield microscope. The scoring of histopathology slides is currently very qualitative and approximate. Our goal is to enable objective and quantitative scoring of slides with a particular emphasis on quantifying the distribution of molecular biomarkers that indicate specific disease conditions. For example, estrogen receptor (ER), progesterone receptor (PR), and Ki67 are examples of molecular biomarkers that are important for diagnosing and sub-typing breast cancers. Our method is based on three basic principles: (i) delineate each cell in the image field as accurately as possible; (ii) identify the types of all cells in the field; and (iii) quantify biomarkers on a cell-by-cell basis in a selective manner based on cell type.
In this application, the images are spatially two-dimensional, but much richer in terms of spectral data per pixel. The specimens are multiplex immunolabeled to highlight key tissue structures of interest (cell nuclei, membranes, cytoplasmic regions of certain cell types, etc.), and the molecular biomarkers of interest. These immunolabeled specimens are subjected to multi-spectral microscopy. The spectral cubes are computationally unmixed to produce a set of non-overlapping channels that are analyzed using the FARSIGHT divide & conquer associative image analysis strategy.
Currently, this system is implemented using a combination of C++ and IDL. It is currently being translated to a C++ and Python based system. The illustrations and instructions below refer to the IDL/C++ system. Users need to download and install the free IDL virtual machine from ITT. This page does require you to register.
Contents |
Background and motivation
Detecting molecules of interest and quantifying them plays an increasingly important role in modern medical diagnosis and treatment planning[1]. Currently, most systems are visually based qualitative descriptors which suffer from the consequent limitations. There are other algorithms that quantify biomarkers on a pixel-by-pixel basis or a regional basis[2-5]. Segmenting the cells and nuclei allows us to classify the different cell types based on antigens that mark specific cell. This enables us to quantify the biomarker of interest in the relevant cell type. This is a more biologically relevant measure and can be more useful in medical diagnosis.
Algorithm
The algorithm involves the following steps:
- Segment(delineate) nuclei
- For this we use the 2-D nucleus segmentation algorithm described here[[1]]
- Delineate cell boundaries
- If reliable membrane or cytoplasm staining is available then:
- Binarize the image by graph cuts[6]
- Create a gradient enhanced distance map with the segmented nuclei as the initialization points ( described in manuscript which will soon be submited )
- Use a seeded watershed algorithm with the gradient weighted map as the feature input and the segmented nuclei as the markers and segment(delineate) the cells[7]
- If neither the cytoplasm nor the membrane staining is reliable then use a purely geometric approach to estimate the cytoplasmic domains:
- Draw dynthetic boundaries by redefining the dominance region described by Nath,et. al.[8] as
- Draw dynthetic boundaries by redefining the dominance region described by Nath,et. al.[8] as
- If reliable membrane or cytoplasm staining is available then:
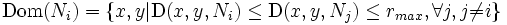
- where (x,y)is any point in the neighborhood of the nucleus Ni and rmax is the user defined maximum radius of of the synthetic boundaries. rmax is in terms of the number of pixels from nucleus Ni
- Quantify biomarker in the region of interest
- Load training set and classify cells as biomarker positive or negative using a Bayesian classifier
Histopathology Program in c++
The histopathology tool in Farsight inherits all the GUI capabilities of the Nucleus Editor and extends the functionality for use in cytometric analysis of histopathology samples. For efficient editing of nuclear segmentation and to use all the functional capabilities of the GUI, read the Nucleus Editor page and then proceed with this page. The work-flow for the cytometric quantification of biomarkers and classification of cells is divided into five steps.
- Load and pre-process channels
- Nucleus Segmentation
- Whole Cell Segmentation
- Computing Associations
- Training and Classification
Load and pre-process channels
Images can be loaded as is from the hard disk. The supported file formats have the following extensions .tif, .pic, .png, .jpg and .lsm Since there is no open source format with an accompanying tool with which a user can conveniently synthesize images composed of channels multiple channels and append meta-data, the histopathology tool includes a method to load channels and meta-data specified in a xml file(hotlink to scr cap of xml). The full path(hotlink to scr cap of xml) of each image has to be specified here(hotlink to scr cap of xml). The display color-space to which the image is rescaled is specified as <r="___" g="___" b="___">(hotlink to scr cap of xml) along with the channel name. The rage for each color is between 0 and 255. If r=g=b=255 the image will be loaded with its native color range but such images can not be used in any processing step. The conversion and rescaling of RGB images loaded as single channels is done internally, so, the format of the image is not a factor. The tag <chname="___">(hotlink to scr cap of xml) specifies the channel name which is used to identify the cahnnel in the processing step. Caution: The case of channel name strings have to match in both the image file and the project definition file. [[Image::Load_multi_channel_image.jpg |thumb|600px|Click on the image to enlarge|center]]
Nucleus Segmentation
Whole Cell Segmentation
Computing Associations
Training and Classification
Histopathology Program in the IDL environment
The algorithms have been implemented with a custom GUI built on IDL and c++. To run the program, you will need the IDL virtual machine which is available online for free[[2]]. Soon, the GUI code will use the QT & VTK Development Framework and added to the Farsight repository.
Steps for analyzing 2-D multi-spectral images of Hisopathology samples
1- Read image channels one-by-one (Read menu):
-Choose "start/end read image channels" -start reading the channels one by one (nuclei, cytoplasm and/or membrane channels) -Choose "start/end read image channels" again

2- Start nuclear segmentation (nuclear segmentation menu):
-Change the nuclear segmentation settings (if needed) -Run initial segmentation -Edit the nuclear segmentation results (optional). This includes merging, splitting, and deleting nuclei -Finalize segmentation. The segmentation result will be displayed and will be saved into a file as well -If a nuclear segmentation results file is already available, choose "Read Final Segmentation"

3- Start cytoplasm (or membrane) segmentation (cytoplasm segmentation menu):
-Change the cytoplasm segmentation settings (if needed) -Run cytoplasm segmentation

4- Quantify the biomarker (feature extraction menu):
-Choose "Quantify Biomarker"
-A new window will then allow you to brows for the biomarker channel (image)
-Set the quantification options:
-Define the region of interest
-Choose whether or not you need to do background subtraction (two- or three-level)
-Quantification results will be saved into a csv file

3- Classify cells (cell classification menu):
-Define classes -Select training samples manually or read them from a file that was saved after previous manual selection of training samples -Train the classifier -Classify the cells -classification results will be displayed (by changing the seeds colors) and will be also saved into a csv file

References
- Hammerschmied CG, Walter B, Hartmann A. [Renal cell carcinoma 2008 : Histopathology, molecular genetics and new therapeutic options.]. Pathologe 2008;29(5):354-63.}
- Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med 2002;8(11):1323-7.
- Mulrane L, Rexhepaj E, Penney S, Callanan JJ, Gallagher WM. Automated image analysis in histopathology: a valuable tool in medical diagnostics. Expert Rev Mol Diagn 2008;8(6):707-25.
- Taylor CR, Levenson RM. Quantification of immunohistochemistry--issues concerning methods, utility and semiquantitative assessment II. Histopathology 2006;49(4):411-24.
- Tamai S. Expert systems and automatic diagnostic systems in histopathology--a review. Rinsho Byori 1999;47(2):126-31.
- Al-Kofahi Y, Lassoued W, Lee W, Roysam B. Improved Automatic Detection & Segmentation of Cell Nuclei in Histopathology Images. IEEE Trans Biomed Eng 2009 (in review)
- Beare R., Lehmann G. The watershed transform in ITK - discussion and new developments. The Insight Journal - 2006 January - June [[3]]
- Nath SK, Palaniappan K, Bunyak F. Accurate Spatial Neighborhood Relationships for Arbitrarily-shaped Objects using Hamilton-Jacobi GVD. Lect Notes Comput Sci 2007;4522 LNCS:421-431.